- Studio Aegle
- Posts
- La psilocibina potrebbe diventare un farmaco a breve
La psilocibina potrebbe diventare un farmaco a breve
Nuovi risultati dallo studio di fase 3 della Compass Pathways
Studio Aegle
21 febbraio 2026
Oggi approfondiamo la notizia dell’anno, e non siamo nemmeno a marzo 😆
Mettiamoci comodi va.
Compass Pathways ha appena reso noti i risultati topline della sua seconda fase 3 sulla psilocibina nella depressione resistente al trattamento.
581 pazienti, età media intorno ai quarant'anni, oltre l'85% caucasica, pochissima esperienza psichedelica pregressa.
Il protocollo prevedeva due dosi da 25 mg a distanza di tre settimane, confrontate con 10 mg e con 1 mg.
A sei settimane, la differenza media della scala MADRS tra 25 mg e 1 mg è stata di 3,8 punti. Endpoint primario raggiunto.
Fin qui, la notizia.
Adesso passiamo al vero significato. Serve un bel grafico di aiuto perché non è proprio intuitiva la spiegazione.
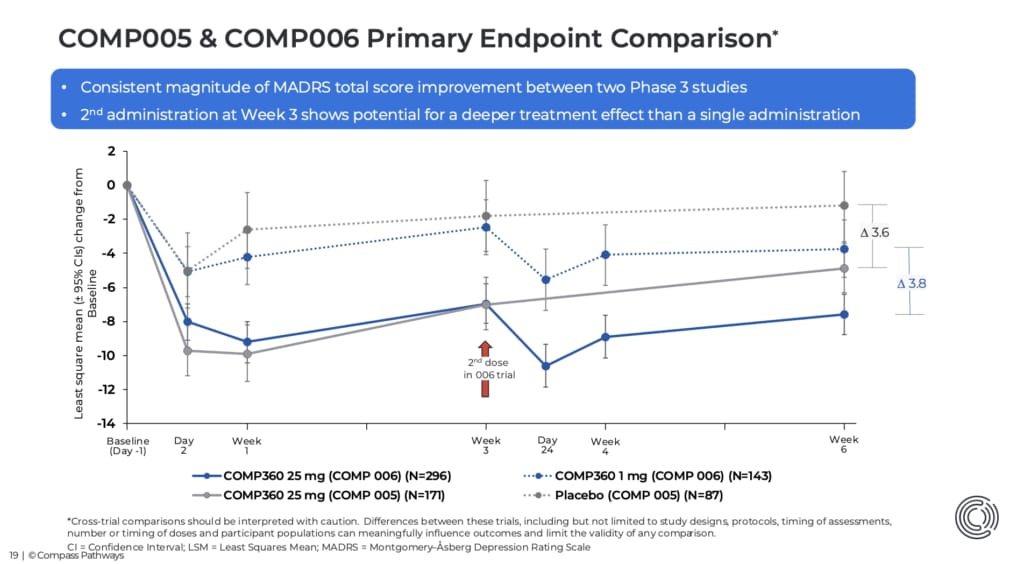
Nella prima fase 3 (COMP005), il confronto era con un placebo inerte (ne ho parlato in questa precedente newsletter). La differenza tra 25 mg e placebo era stata di 3,6 punti e il placebo era migliorato di poco più di 1 punto rispetto al basale.
In COMP006 il confronto è stato con 1 mg di psilocibina, e 1 mg non è un placebo vuoto: non produce un'esperienza psichedelica piena, ma qualcosa fa (la tifoseria del microdosing sta facendo standing ovation, vi vedo). Lo mostrano i dati: il gruppo da 1 mg migliora di quasi 4 punti dal basale, non solo 1 punto come era successo con il placebo.
Il gruppo da 25 mg, dal canto suo, migliora di circa 7,6 punti, più che nella prima fase 3. La seconda dose incrementa ulteriormente la risposta. Ma siccome anche il gruppo di confronto migliora molto di più rispetto al placebo inerte di prima, la distanza finale tra i due rimane quasi identica: 3,8 contro 3,6.
È come se in una gara di salto in alto entrambi gli atleti migliorassero di molto rispetto alla competizione precedente. Il campione salta più in alto di prima. Ma anche lo sfidante salta molto più in alto. Il risultato finale? Il distacco tra i due rimane quasi lo stesso.
Questo è il nodo, ed è qui che Compass e gli analisti si guardano storto.
L'azienda enfatizza la riduzione assoluta “Oh guardate quanto migliorano i pazienti trattati!!!”, e ha ragione a farlo, perché quel numero è più grande.
Gli analisti invece guardano la distanza tra i gruppi, che è quella che conta per dimostrare l'efficacia differenziale e dicono “Mah, ci pare che due dosi abbiano la stessa efficacia di una”.
Entrambi leggono gli stessi dati. Nessuno dei due mente. Ma la verità è che stanno rispondendo a due domande diverse…
C'è poi il tema dei 10 mg, che merita una menzione a parte.
Il gruppo da 10 mg ha mostrato un effetto sorprendentemente vicino a quello da 25 mg in diversi time point e tutto ciò è inatteso, perché nella fase 2b precedente il 10 mg non aveva convinto per niente. Goodwin stesso, il Chief Medical Officer di Compass, non ha una spiegazione definitiva (per la sincerità dimostrata ha guadagnato punti simpatia).
Ma se il 10 mg ha funzionato quasi quanto il 25 mg, le implicazioni sul profilo rischio-beneficio (un'esperienza meno intensa, una logistica più semplice) non sono banali. Da tenere d'occhio per il futuro.
Un'ultima cosa sui numeri, prima di passare oltre. Nelle fase 3 compare una categoria chiamata "riduzione clinicamente significativa", definita come almeno il 25% di miglioramento del punteggio. Nella letteratura standard il criterio di risposta è però ufficialmente il 50%.
La giustificazione non è irragionevole, Compass ci dice che in una popolazione resistente al trattamento anche il 25% di miglioramento può fare la differenza. Una scoperta sensazionale, senza dubbio, meno male che ce lo hanno spiegato loro, ma forse questa coscienza da buon samaritano che è spuntata all’improvviso ha un secondo fine?
Abbassare la soglia significa anche che le percentuali di "migliorati" crescono automaticamente. Quando si legge che i pazienti hanno raggiunto una riduzione clinicamente significativa, la valutazione va fatta con cautela. Quindi quando Compass ci dice che "oltre il 40% dei partial responders va in remissione", in termini assoluti si tratta di poco più di una dozzina di persone. Non è falso. Ma va messo nella giusta proporzione, abbassiamo l’hype.
Sul fronte sicurezza, non emergono nuovi segnali… o sì??? 👀👀👀
Teniamoci forte, ma forte forte eh.
Le allucinazioni visive compaiono per la prima volta come evento avverso registrato.
No Maria, io esco.
Goodwin ha ammesso con una certa onestà che classificare come "effetto avverso" ciò che costituisce in buona parte l'effetto principale del farmaco non è una scienza esatta. È uno di quei momenti in cui il linguaggio regolatorio e l'esperienza soggettiva si guardano in modo un po' imbarazzato, senza sapere bene cosa dirsi. Goodwin, che non mi è mai andato particolarmente a genio, continua a guadagnare punti simpatia con questa fase 3.
Il parallelismo con l’esketamina è immediato, dove la dissociazione viene considerata un effetto collaterale del trattamento quando è il trattamento stesso.
La spiegazione ufficiale di questa supercazzola è in realtà tecnica: con 96 centri e criteri standardizzati, ogni “sintomo” (sia positivo che negativo - un caro saluto a MAPS che è stata cazziata dalla FDA per non aver riportato i sintomi positivi) finisce nel sistema di raccolta dati.
C'è però una lettura meno innocente: classificare le allucinazioni visive come effetto collaterale è una mossa regolatoria molto comoda. L'FDA sa gestire un effetto avverso noto. Sa molto meno come gestire un'esperienza che è al tempo stesso il rischio e il meccanismo terapeutico. Compass sposta così l’eventuale responsabilità sul clinico che non ha saputo gestire un rischio dichiarato, e soprattutto rafforza la narrativa del farmaco puro. Chapeau.
Poi arriviamo alla parte davvero scivolosa. La psicoterapia.
Di nuovo, il caro Goodwin è stato esplicito: “abbiamo lavorato duramente per minimizzare la preparazione e il supporto psicologico”. Ottimo… ci fa proprio tanto piacere.
L'obiettivo era dimostrare un effetto farmacologico puro, isolare la molecola dal contesto. È comprensibile, l'FDA approva farmaci, non scuole di psicoterapia (di certo non ha approvato il manuale di psicoterapia della MDMA-AT). Per ottenere un'indicazione bisogna dimostrare che la sostanza di per sé produce un effetto misurabile.
Qui nasce il paradosso, l’ennesimo.
La terapia psichedelica ha sempre considerato preparazione e integrazione come pilastri centrali. Non optional. Ma Levine, Chief Patient Officer, stupisce davvero con effetti speciali quando se ne è uscito con la perla:
“I think we’re making donuts, and we will put them out into the world, and healthcare providers will put the icing on them and the sprinkles”
Non sta dicendo che la psicoterapia è inutile, ci sta dicendo che è molto utile, solo la farà qualcun altro al posto di Compass.

Se nel trial fai il minimo indispensabile per soddisfare i requisiti regolatori, e poi suggerisci che nella pratica clinica i terapeuti potranno aggiungere ciò che è stato tolto, stai spostando il peso terapeutico fuori dallo studio. E allora la domanda diventa inevitabile: chi stabilisce cosa sia un'integrazione adeguata? Ogni centro farà la sua? Ogni clinico il suo modello?
Dal punto di vista clinico (e dei fuffaguru) è una terra fertile. Dal punto di vista della ricerca è un buco enorme. Perché se l'integrazione è davvero determinante per l'esito a lungo termine, e non la studiamo in modo rigoroso, stiamo approvando una molecola su dati che raccontano solo metà della storia.
Il risultato è che i numeri che abbiamo oggi, pur positivi, non sono rivoluzionari. Sono compatibili con un'approvazione. Sono coerenti. Sono replicati su più di mille pazienti in tre studi. Ma non raccontano una terapia che ribalta il paradigma della psichiatria.
Il paradosso è proprio questo: per far entrare la psilocibina nel sistema, bisogna trattarla come un qualsiasi altro farmaco. Ma trattandola come un qualsiasi altro farmaco, si rischia di amputare la parte che l'ha resa interessante fin dall'inizio.
Compass deve presentare un NDA, puntare a una priority review, arrivare a un possibile lancio nel 2027. È un'azienda quotata, con investitori, con una traiettoria commerciale da difendere. Tutto questo è reale. Ma altrettanto reale è la tensione tra ciò che è approvabile e ciò che è terapeuticamente sensato.
Compass ha letto la bocciatura della MDMA-AT come un manuale di istruzion e ci ha costruito sopra una strategia vincente.
Forse il vero punto non è se 3,8 è tanto o poco. Forse il vero punto è che la ricerca psichedelica, arrivata alla soglia dell'approvazione, è costretta a scegliere cosa lasciare fuori dal modello per poter entrare nel sistema.
Quello che ha deciso di lasciare fuori potrebbe essere proprio la parte più trasformativa.
È questa la zona che mi affascina. Non la vittoria o la sconfitta di un titolo in borsa. Ma il modo in cui la regolazione, la metodologia e la clinica si intrecciano e si contraddicono.
Perché la psilocibina sta per diventare, forse, un farmaco approvato.
Ma la terapia psichedelica è un'altra cosa.
Alla prossima! 😎
Non sei ancora iscritto alla newsletter?